Track Your Plaque

How to Reverse Heart Disease with the Coronary Calcium Score
by Jeffrey Dach MD
Finally Accepted by the AHA
The AHA (American Heart Association) has steadfastly denied for many years that Coronary Calcium Scoring was a valid marker of heart disease. Well guess what? They have recanted, and admitted that the amount of calcium in the coronary arteries reliably predicts heart attack risk. This is called the calcium score.(1)
Dr Matt Budoff
UCLA cardiologist, Dr. Matt Budoff, a long-time champion of the Coronary Calcium Scan, and author of the AHA paper says, “The total amount of coronary calcium (Agatston score) predicts coronary disease events beyond standard risk factors.”(1)
Image upper left, courtesy of Wikipedia Rembrandt, The Anatomy Lesson.
Dr. Detrano
Dr. Detrano, in a recent article in NEJM (New England of Medicine), confirms that “The coronary calcium score is a strong predictor of incident coronary heart disease and provides predictive information beyond that provided by standard risk factors”. (31) The Coronary Calcium Score is a precise quantitative tool for measuring and tracking heart disease risk, and is more valuable and accurate than other traditional markers (such as total cholesterol which is practically worthless as a heart disease risk marker).
What is Coronary Artery Disease? It’s Plaque Formation.
Coronary Arteries in Cross Section

Age 20-30 years Age 50-70 years
In youth, at left, there is minimal plaque formation. However, at right with passage of time the plaque grows larger. About 20% of this plaque volume contains calcium which is measurable on CAT scan, providing a marker for the total plaque burden. Calcium score and by inference, plaque volume typically increases 30-35% per year in untreated patients.
Note that even though the right vessel has a larger plaque, the lumen has remodeled so that the inner diameter remains freely open. Eventually, as we age, the enlarging plaque eventually obstructs blood flow causing a heart attack. Another common scenario is plaque rupture which exposes the inflammatory debris of the plaque to the circulating blood. This quickly results in clot formation (thrombosis) resulting in a heart attack and possibly sudden death.
Repeat: The calcified portion of the plaque is consistently 20% of the total plaque volume, allowing use of the calcium score as a marker for total plaque volume.
Arterial Calcification – Why Does it Happen?Image below: Microscopic view of arterial calcification
(yellow arrows outline blue calcifications)

Calcification in the soft tissues (connective tissue, ligaments, muscles, arteries) is found in many disease states, and commonly identified on pathology slides of tissues. Whenever there is cell death or tissue necrosis (death of cells), the body invokes a process of calcification which can be regarded as part of the healing process. Arterial calcification is actually a form of bone formation in the wall of the artery triggered by an inflammatory process. Pathology studies have shown that coronary artery calcium forms in areas of healed plaque ruptures. (21) Calcification and plaque formation increases with age, with calcium score typically increasing 30-35% per year in untreated patients (William Davis MD).
Heart Attack Rate Associated with Calcium Score
Chart below shows increasing heart attack rate as coronary calcium goes up

Chart Above shows that Calcium Score is Highly Predictive of Heart Attack Risk
What is a Heart Attack?
 A heart attack is cell death of heart muscle caused by lack of blood flow with oxygen deprivation. AS previously mentioned, this is caused by a arterial blockage by enlarging plaque formation which occludes the lumen, or plaque rupture which causes clot formation which occludes blood flow. If a small area of heart muscle is involved, the heart attack may be silent with no symptoms. If a large area is involved, there may be severe chest pain radiating to the left arm or jaw, or other symptoms such as shortness of breath. If the conduction system is involved, there may be irregular heart rhythm called ventricular tachycardia which can cause sudden death. Some people have chronic chest pain from diseased arteries and this is called angina pectoris, treated with medicines to dilate the arteries such as nitroglycerine.
A heart attack is cell death of heart muscle caused by lack of blood flow with oxygen deprivation. AS previously mentioned, this is caused by a arterial blockage by enlarging plaque formation which occludes the lumen, or plaque rupture which causes clot formation which occludes blood flow. If a small area of heart muscle is involved, the heart attack may be silent with no symptoms. If a large area is involved, there may be severe chest pain radiating to the left arm or jaw, or other symptoms such as shortness of breath. If the conduction system is involved, there may be irregular heart rhythm called ventricular tachycardia which can cause sudden death. Some people have chronic chest pain from diseased arteries and this is called angina pectoris, treated with medicines to dilate the arteries such as nitroglycerine.
Common Sites of Plaque Formation – Bifurcations and Mechanical Stress


(image at right courtesy of Wikipedia)Above Left image shows xray angiogram of typical ulcerated plaque with stenosis at carotid bifurcation, Above Right Image shows gross pathology of inside of the vessel with darkened plaque (arrow). In this example, we have an artery in the neck that feeds blood flow to the brain. Plaque rupture and occlusion of the artery in this case caused a stroke, however, the same process occuring in the heart causes a heart attack.
Branching Vessels – Increased Turbulence
Ask any interventional radiologist or invasive cardiologist where they find the plaque formation and obstructions in the arterial tree, and they will say its the same few places over and over again. These places are the carotid bifurcation, the distal aorta at the bifurcation, the femoral bifurcation, the exit from the adductor canal. And of course, the proximal coronary arteries, and bifurcations of the coronary arteries. A birfurcation is where the vessel branches into two vessels, making a Y pattern.
 The bifurcations have maximal turbulance and mechanical stress on the vessel wall. Remember the blood is flowing under pulsatile pressure, and this mechanical pressure and turbulence, over time, causes little stress cracks in the vessel. The cracks appear at sites of maximal stress. The coronary arteries are a special case because of the extra motion of the cardiac muscle which moves and stretches the coronary arteries every heart beat, especially as the arteries branch off from the aorta which is relatively stationary, while lower down over the surface of the heart, the vessels move vigorously with each heart beat. Atherosclerosis is essentially the net result of the healing process for these little cracks in the arterial wall resulting from mechanical stress.
The bifurcations have maximal turbulance and mechanical stress on the vessel wall. Remember the blood is flowing under pulsatile pressure, and this mechanical pressure and turbulence, over time, causes little stress cracks in the vessel. The cracks appear at sites of maximal stress. The coronary arteries are a special case because of the extra motion of the cardiac muscle which moves and stretches the coronary arteries every heart beat, especially as the arteries branch off from the aorta which is relatively stationary, while lower down over the surface of the heart, the vessels move vigorously with each heart beat. Atherosclerosis is essentially the net result of the healing process for these little cracks in the arterial wall resulting from mechanical stress.
 William Davis MD
William Davis MD
Advocate of the Coronary Calcium Score
William Davis MD recommends screening CAT heart scans in males over 40 and females over 50. He would start at younger ages if high-risk features are present, such as strong family history of early heart disease, cigarette smoker, diabetes mellitus, or severe lipid or lipoprotein genetic disorders. (2)(3) The Coronary Calcium Score test is currently covered by Medicare and many health insurances.(32)
Credit and Thanks is given to William Davis MD at the Track Your Plaque Web site for much of the information in this article. I have added and embellished some of the information.
What does Calcium Look Like?Normal Calcified


Calcium looks White on the CAT scan (red arrows at right).
(Above) CAT scans of the Coronary Arteries.
The left image shows a normal coronary artery (red arrows), while the right image shows a heavily calcified coronary artery (white line outlined by red arrows) indicating high risk for coronary artery disease and heart attack.
All About Coronary Calcium Scoring
 1) Calcium scoring may be superior to angiography as a means to track plaque. That’s because the vast majority of heart attacks are due to plaque rupture and thrombosis at areas of thickened plaque with minimal lumen narrowing. Over time, the body’s healing process automatically remodels the areas of thickened plaque, and increases lumen size to compensate for the reduced blood flow.
1) Calcium scoring may be superior to angiography as a means to track plaque. That’s because the vast majority of heart attacks are due to plaque rupture and thrombosis at areas of thickened plaque with minimal lumen narrowing. Over time, the body’s healing process automatically remodels the areas of thickened plaque, and increases lumen size to compensate for the reduced blood flow.
2) Calcium scoring gives a precise number which correlates with the amount of plaque volume. Although only the hard plaque, or calcium in the artery is actually measured, this is useful because it consistently occupies 20% of plaque volume (Total hard and soft plaque).
3) The new 64-slice CAT scanners provide reliable calcium scoring just like any other scanner, both multi-slice and EBT(Electron Beam Cat).
 The Track Your Plaque Program, by William Davis MD(2)(3)
The Track Your Plaque Program, by William Davis MD(2)(3)
1) Quantify plaque with Coronary Calcium Score with CAT scan (or with Electron Beam CT). Obtain your CAT Scan serially, every 12 months to assess response to treatment and lifestyle modification (track your plaque).
2) Use Sophisticated Lipoprotein Panel (Quest-Cario-IQ , LabCorp-VAP)(7-8)) to uncover hidden causes of plaque progression. LDL particle size and number, Lipoprotein (a). Repeat every 6 months.
3) The Main Treatment Goal is the reduction in Coronary Artery Calcium Score, and by inference, reduction in plaque volume and reduction in cardiovascular mortality. The cardiology community still awaits the hard data on these results (CHD mortality and CHD events, treatment arm vs no treatment arm). These numbers have not been published as far as I know.
How to Measure Success in Halting or Reversing Heart Disease Plaque
According to Dr. Davis, calcium score typically increases at an astonishing rate of 30-35% per year without treatment. Therefore, Dr. Davis considers treatment success to be reduction in this rate from 30 to perhaps only a 5-10 per cent increase in calcium score per year. An absolute reduction in calcium score on follow up scanning is the optimal outcome, which is difficult to achieve even with strict adherance to the Track Your Plaque program, in Dr Davis’s experience.
Track Your Plaque Program Details – Attain the Following Targets:
a) Reduction of LDL to 60 mg/dl (LDL should be measured directly, not calculated)
b) Reduction of triglycerides to 60 mg/dl.
c) Raising HDL to 60 mg/dl.
d) Correction of hidden causes of plaque on Lipoprotein profile such as total number of small LDL particles, IDL, and Lp(a).
e) Achieving normal blood pressure (<130/80) Even a small elevation of blood pressure in diseased arteries can cause increased mortality. Diseased arteries are fragile and plaque rupture can occur easily.
f) Achieving normal blood sugar (=100 mg/dl). Diabetes is a high risk factor for heart disease.
g) Reduction of C-reactive protein to <1 mg/l
Dietary Modification and Supplements to Attain Above Targets:
Niacin
a) Niacin vitamin B3 (Slo-Niacin Upsher-Smith (44) or Niaspan Kos Pharmaceuticals preferred) 500-1500mg. per day (avoid the no-flush niacin which contains inositol).(6)(44)
Buy Niacin
on Amazon.
Omega 3 Fish Oil
b) Fish oil (Omega 3 oils) 4000 mg per day (providing 1200 mg omega-3 fatty acids). (molecular distilled pharmaceutical grade).(36)
Buy Purecaps Super-Critical Co2 Extract Omega 3 Fish Oil
Vitamin D
Vitamin D level restored to above 50 ng/ml (Vitamin D3 2000-5,000 u/day), Vitamin K2 also used. Low vitamin D is associated with increasing arterial calcification and increased heart disease risk. (26)Consumption of calcium tablets by women increases arterial calcification and heart attack risk.(5)
Buy Vitamin D
Read my article on vitamin D which can be found here.(60)
d) Low Glycemic Diet (avoid Fructose Corn Syrup, avoid wheat products), and eliminate wheat products like Shredded Wheat cereal, Raisin Bran, and whole wheat bagels.
e) Consume foods such as raw almonds, walnuts, pecans; olive oil and canola oil. Beneficial for lipoprotein profile.
f) Increasing protein intake, our major building block for body tissues. Added benefit of protein intake is that it doesn’t increase blood sugar. This is low glycemic nutrition.
g) Wine—Red wines contain resveratrol, (don’t exceed two glasses/ day). Bioflavonoids and anti-oxidants have a strong anti-inflammatory effect.
h) Fiber – Gound flaxseed (2 tbsp/day)-Extra fiber aids in detoxifying liver and the entire body by interrupting the enterohepatic circulation. Psyllium (metamucil). Regulates bowel movements and has favorable effect on lipoprotein profile.
Vitamin C – A Genetic Deficiency Disease
Vitamin C (1000–3000 mg/day), is a key player, as it is the vitamin for strong collagen formation, strengthening the arterial wall. See Linus Pauling’s patented protocol which includes Vitamin C and amino acids Proline and Lysine, the two amino acids that act as receptors for Lp(a). By consuming additional Lysine and Proline, the receptor sites on the Lp(a) and other lipoproteins are covered up and made less sticky, resulting in less deposition in the artery wall. The vitamin C is important not only for strong collagen formation, a major component of the arterial wall, but also for all other structural elements of the body, for that matter. (37)(52)(53)(54)(55)(56)(57)
Humans have a genetic deficiency in Gulano-Lactone-Oxidase (GLO), the final enzyme step in the manufacture of Vitamin C, and therefore unlike all the other animals who make their own Vitamin C, we cannot make this necessary vitamin. We share with all other primates this genetic disease, the inability to manufacture vitamin C, producing a vitamin C deficiency state in all humans.(58)
Also see Thomas Levy’s two books on Vitamin C. (49-51)
Buy Vitamin C at Tower Labs – Ascorcine a9
j) Exercise and weight loss- improves insulin sensitivity, reduces inflammatory markers, reduces blood pressure, improves lipoprotein profile.
Magnesium
k) Magnesium supplementation is inexpensive and safe. Magnesium deficiency due to dietary deficiency or thiazide diuretics for hypertension is common, and is associated increased heart disease risk. Magnesium reduces blood pressure, relaxes smooth muscle in arteries, and is needed for normal endothelial function.(41-43)
Buy Magnesium
L-Arginine
L-arginine is converted to nitric oxide, an important substance for arterial health. Research by Furchgott and other showed that nitric oxide (NO) relaxes arterial smooth muscle, dilating coronary arteries by up to 50%.(35) However, Nitric Oxide (NO) is gone after a few seconds, so it must be replenished at a constant rate to keep the arteries relaxed and open. Lack of NO is associated with constricted arteries, damage to the arterial lining, and accelerated plaque growth. L-arginine shrinks coronary plaque, corrects “endothelial dysfunction”, improves insulin sensitivity, is anti-inflammatory and shrinks plaque. Dosage: l-arginine 6000 mg twice a day, best taken on an empty stomach.
Buy Arginine
Reverse Cholesterol Transport and Essential Phospholipid – Phosphatidyl Choline (PC) (38)(39)
James C. Roberts MD FACC, a practicing cardiologist, lectures extensively on his clinical success with Phosphatidylcholine (IV or in Liposomal Oral Format with EDTA): Reverse Cholesterol Transport and Metal Detoxification. A DVD of his lectures is available which describes considerable clinical success with oral EDTA and phosphytidylcholine. This page contains his DVD lecture material complete with clinical case histories.(61)
Essential Phospholipid is available under trade name Phoschol from Nutrasol which increases Lecithin Cholesterol Acyl Transferase activity (LCAT) (Dobiasova M 1988).(40)(40b)
Activating LCAT is beneficial because LCAT is the crucial substance which transports cholesterol from the arterial plaque back to the liver for metabolic breakdown into bile. This process reverses atherosclerotic plaque formation. Usual Dosage is 3 softgels Nutrasol Phoschol a day each containing 900 mg Phosphatidyl Choline.(38)(39)
Berberine
Berberine, also called Oregon Grape, is a botanical which has many benefits. Dr Roberts page on Berberine for Preventing and Reversing Heart Disease.
Thyroid Function
Normalize thyroid function. Broda Barnes MD showed that low thyroid function was a significant risk factor for heart disease. This conclusion was based on autopsy data from Graz Austria and detailed in his books, Hypothyroidism the Unsuspected Illness, and his other book, Solved the Riddle of Heart Attacks. Barnes felt that the thyroid lab tests were frequently unreliable, and he used clinical judgement instead. (59)
LipoProtein (a)
All About Reducing Lipoprotein (a)(2)(3)
Lipoprotein little A, also written as Lp(a) is a genetic variant lipoprotein which is associated with a high risk of heart disease, and therefore identification and reduction is essential. The problem is that the conventional Lipid panels done in your doctor’s office do not include Lp(a). Only the more sophisticated lipoprotein panels such as the Cardio-IQ (Quest), VAP (Atherotech) or NMR (Liposcience) panels provide Lp(a) data.
Lp(a) and Lipoproteins:
1) Lp(a) is best to measured in (nmol/l), and target below 75 nmol/l .
2) Lp(a) measured in mg/dl (weight may not be accurate), then target below 30 mg/dl .
3) Measured (not calculated) LDL target 50–60 mg/dl.
4) LDL particle number target (NMR) of 600–700 nmol/l or apoprotein B of 50–60 mg/dl. Reduce small LDL to <10% of total LDL.
Treating Lp(a) with Niacin
Use Niaspan® (Kos Pharmaceuticals) or over-the-counter Slo-Niacin® (Upsher-Smith). Both are better tolerated than OTC plain niacin, which may cause the hot flushes. Reduce hot flushed by drinking a full glass of water with each gelcap, and some find adding an aspirin tablet to the routine helps to reduce flushing.
Lp(a) and BioIdentical Hormones
Bio-Identical hormones are beneficial for reducing heart disease. In menopausal females, estrogen preparations such as Bi-Est are used. Estrogens have been shown to reduce coronary artery calcium score.(46)
In males over 50, bio-identical testosterone cream may lowers Lp(a) by as much as 25% (per William Davis MD). Medical studies show that optimizing Testosterone levels in aging males can reduce risk of coronary artery disease by 60%. (47)(48)
DHEA can promote weight loss, and improve insulin sensitivity.(45)
Lp(a) and L-Carnitine
The supplement L-carnitine can be a useful adjunct; 2000–4000 mg per day (1000 mg twice a day) can reduce Lp(a) 7–8%, and occasionally will reduce it up to 20%.
Buy L-Carnitine
Remember, reduction in calcium score on follow up calcium scan is the goal.
What about Statin-Cholesterol Lowering drugs?
Dr Davis admits that the total cholesterol and the LDL cholesterol numbers are of little value in predicting heart disease risk. And he says that the statin drug side effects, ie. muscle pain and weakness, are more common in actual practice than the drug advertising would suggest, making statin drugs difficult to take for the long term.
In my opinion, statin drugs are not recommended for women as explained in my previous article on Statin Drugs for Women, which can be found here (33)
My other article on Statins, Lipitor and the Dracula of Medical Technology can be found here. (34)
What about Calcium Supplements for women to prevent osteoporosis?
Dr Davis points out that women who take calcium tablets have double the risk of heart attacks than those on placebo.(5)
Check out my earlier Heart Disease Reversal Page here.
Credit and Thanks is given to William Davis MD at the Track Your Plaque Web Site and Blog for the above information.(2)(3)

 190
190
 show that
show that 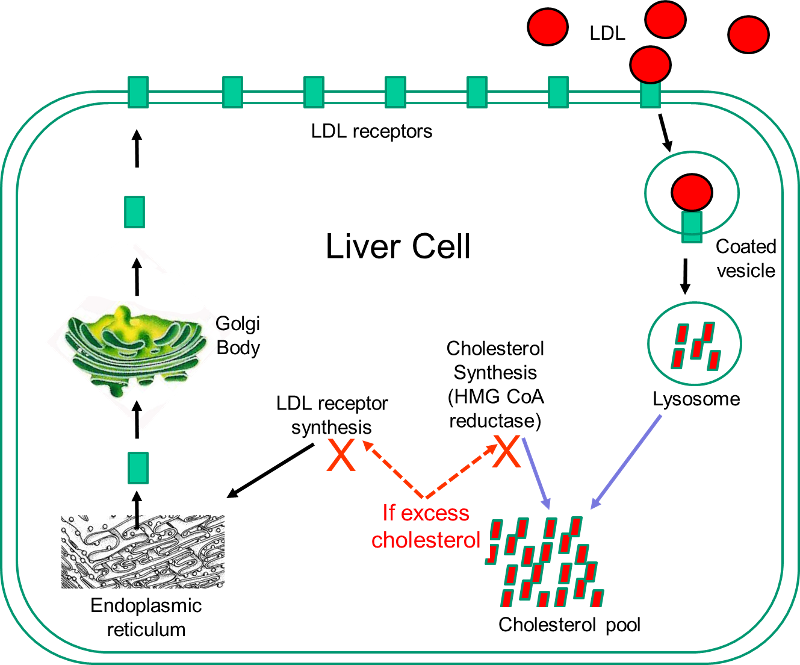 Statins are currently the most powerful cholesterol-lowering drugs available. They act by inhibiting the action of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase, which is the rate-limiting enzyme in the sequence of steps by which cholesterol is synthesized in the liver. The liver has two sources of cholesterol: it can take up LDL particles from the blood, or it can synthesis cholesterol using HMG-CoA reductase. The diagram to the right illustrates cholesterol homeostasis in a liver cell. LDL (shown in red) can bind to LDL receptors on the surface of the liver. Binding causes the LDL to be taken up by the liver cell and digested in a lysosome. Fatty acids and amino acids in the LDL are recycled, and the cholesterol enters the cholesterol pool in the liver cell. Note that if the liver cell synthesizes cholesterol, this too will be added to the cholesterol pool. If the concentration of cholesterol in the liver cells exceeds a certain level, HMG CoA reductase is inhibited, and the synthesis of LDL receptors is also inhibited.
Statins are currently the most powerful cholesterol-lowering drugs available. They act by inhibiting the action of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase, which is the rate-limiting enzyme in the sequence of steps by which cholesterol is synthesized in the liver. The liver has two sources of cholesterol: it can take up LDL particles from the blood, or it can synthesis cholesterol using HMG-CoA reductase. The diagram to the right illustrates cholesterol homeostasis in a liver cell. LDL (shown in red) can bind to LDL receptors on the surface of the liver. Binding causes the LDL to be taken up by the liver cell and digested in a lysosome. Fatty acids and amino acids in the LDL are recycled, and the cholesterol enters the cholesterol pool in the liver cell. Note that if the liver cell synthesizes cholesterol, this too will be added to the cholesterol pool. If the concentration of cholesterol in the liver cells exceeds a certain level, HMG CoA reductase is inhibited, and the synthesis of LDL receptors is also inhibited.





